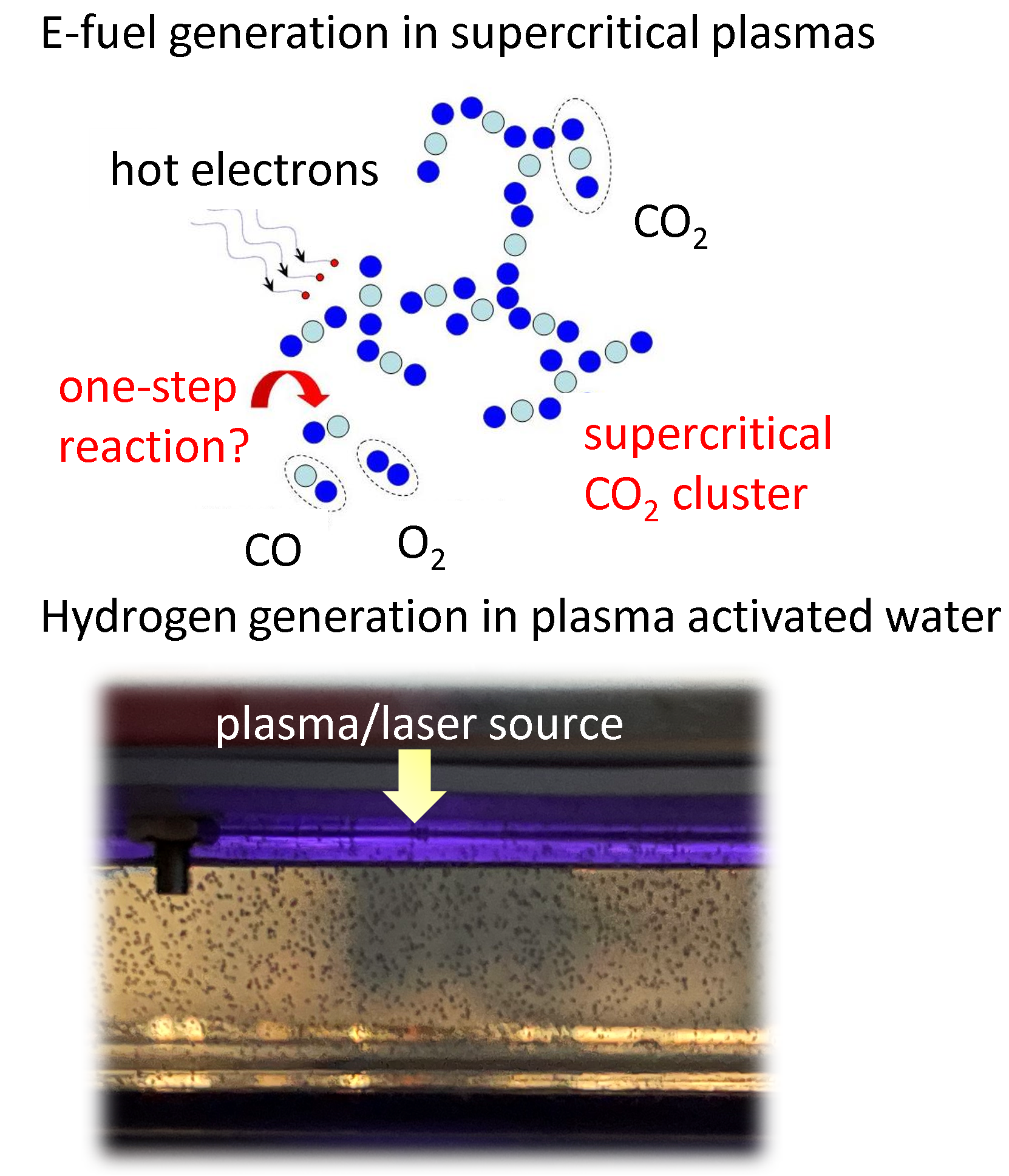
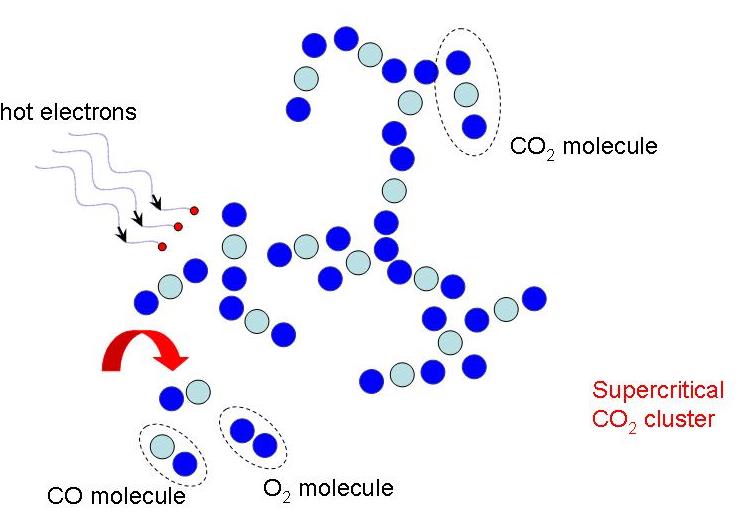
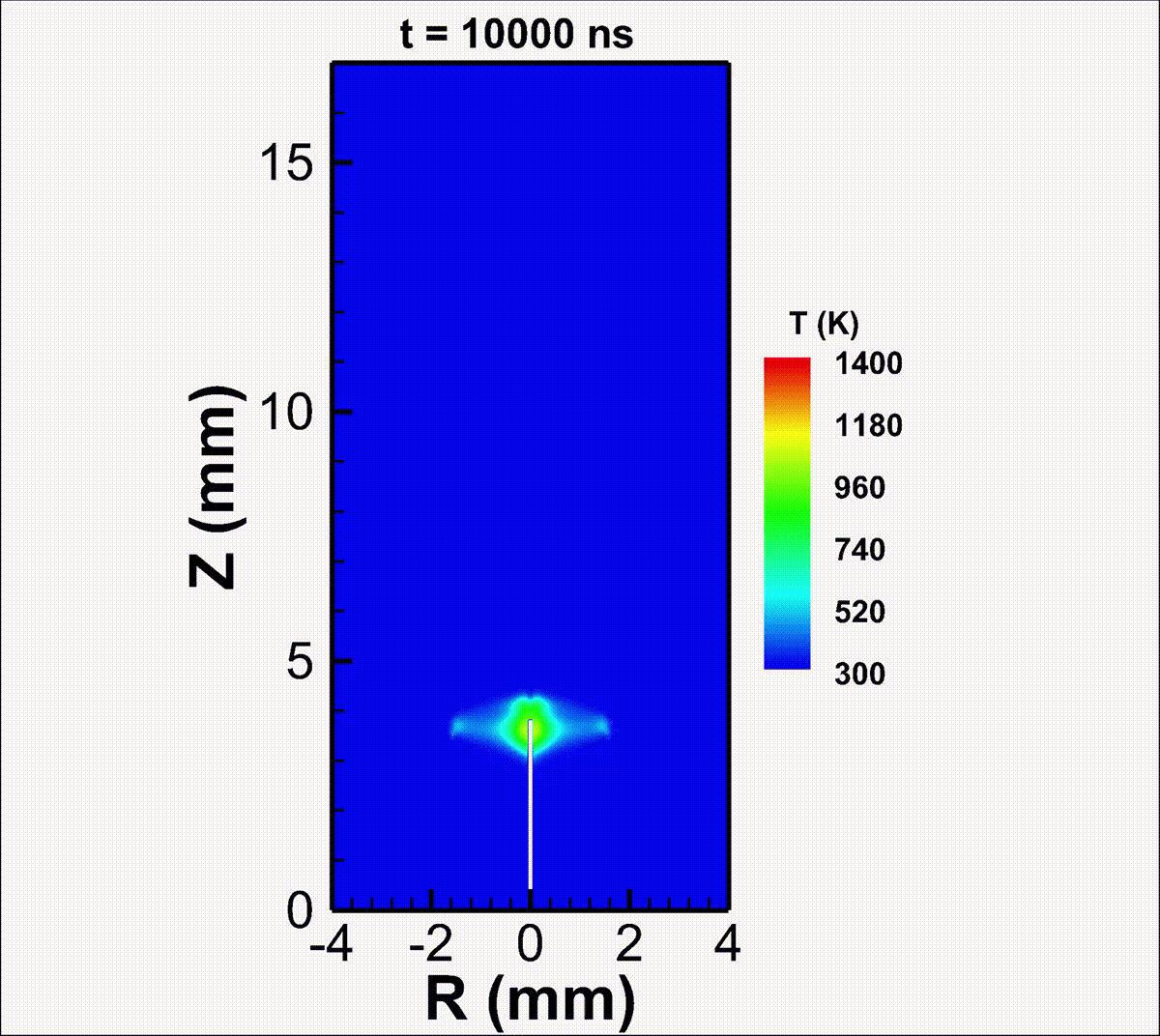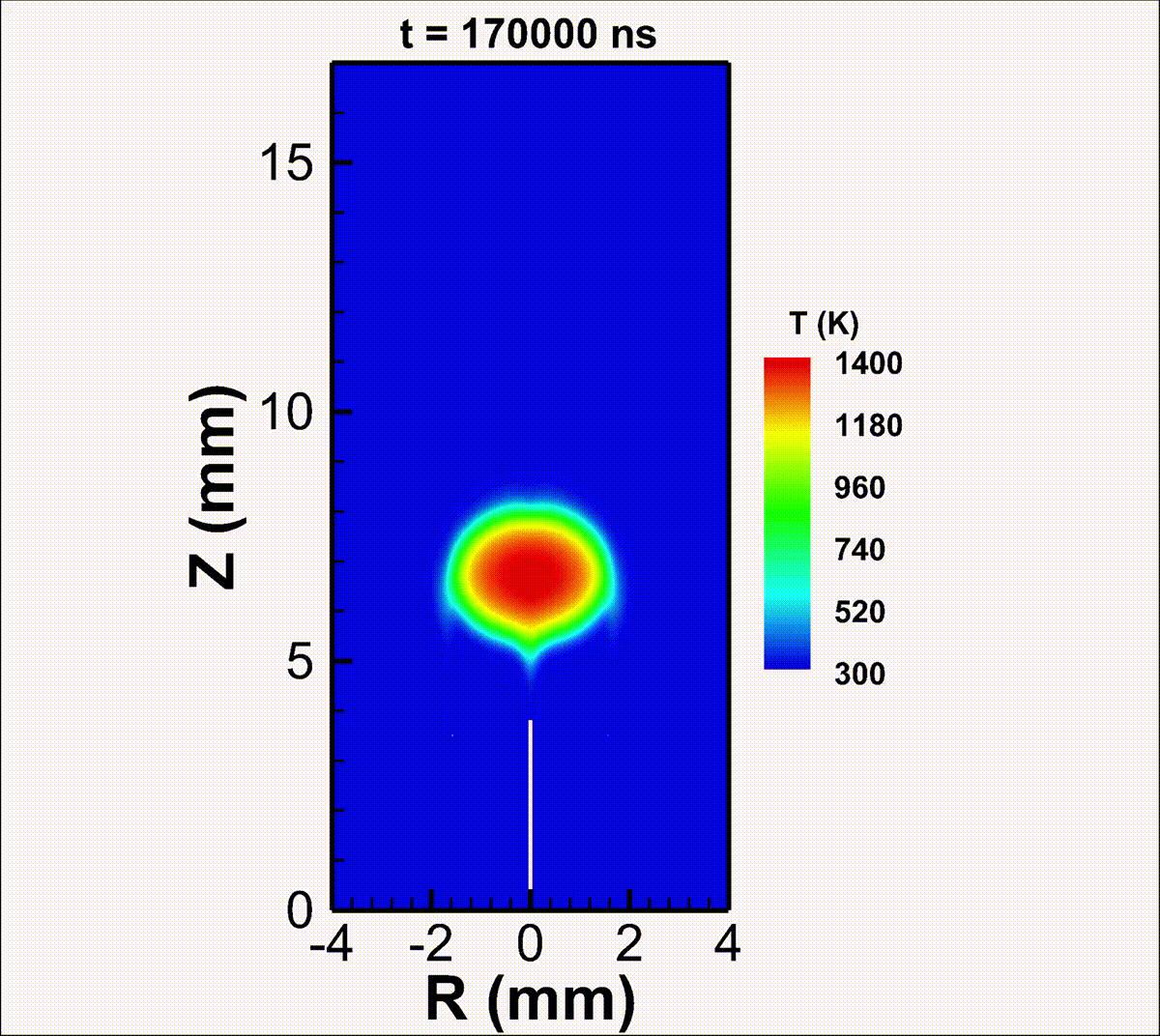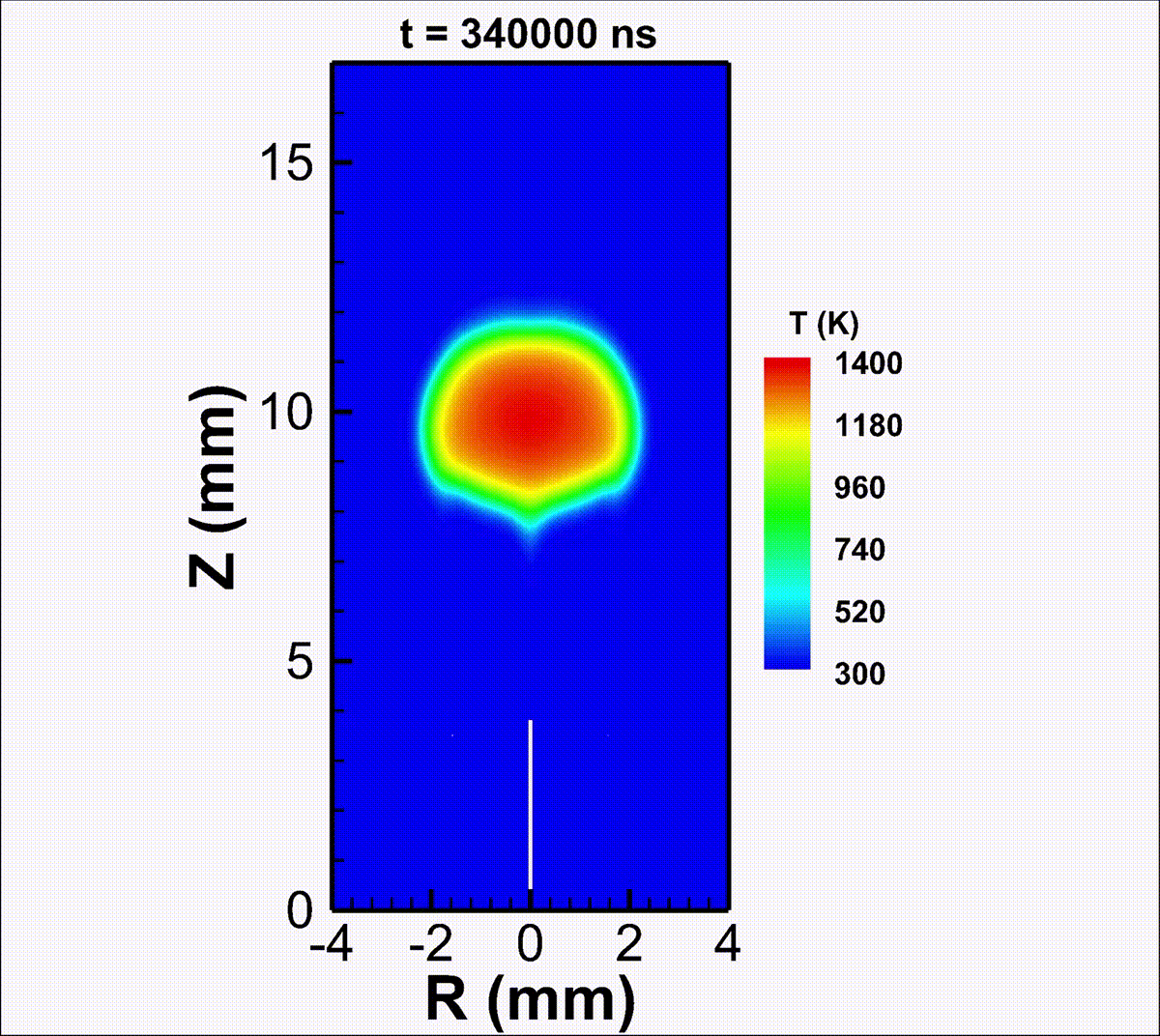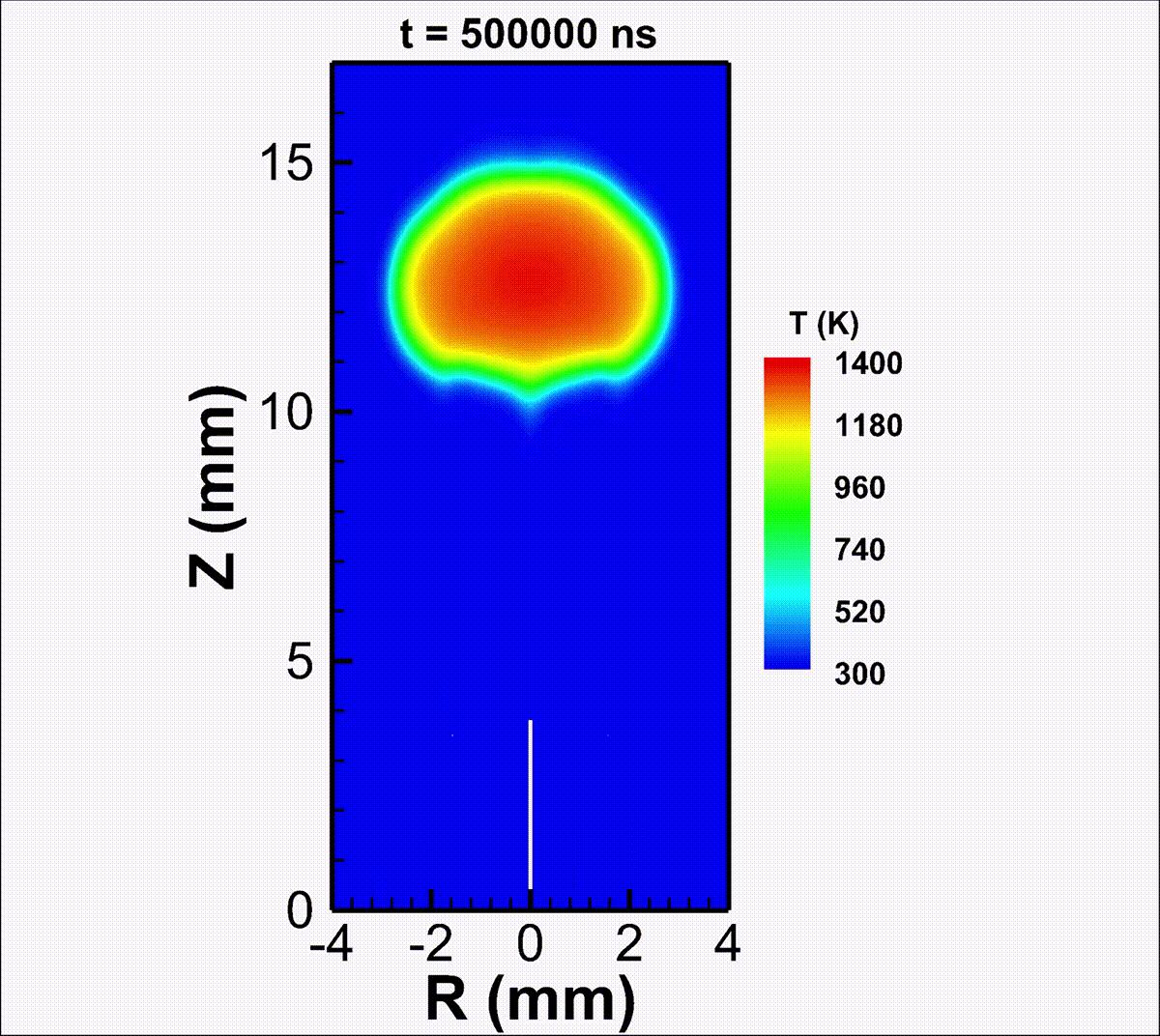
We investigate the conversion of CO2 into CO and O2 with nanosecond repetitively pulsed discharges (NRP) in a high-pressure batch reactor. Stable discharges are obtained at up to 12 bar. By-products are measured with gas chromatography. The energy efficiency is determined for a range of processing times, pulse energy, and fill pressures. It is only weakly sensitive to the plasma operating parameters, i.e., the extent of CO2 conversion is almost linearly-dependent on the specific energy invested. A conversion rate as high as 14% is achieved with an energy efficiency of 23%. For long processing times, a saturation in the yield and a drop in efficiency are observed, due to the increasing role of three-body recombination reactions, as described by zero-dimensional detailed kinetic modeling. The modeling reveals the presence of three-stage kinetics between NRP pulses, controlled by electron-impact CO2 dissociation, vibrational relaxation, and neutral elementary kinetics. Transport effects are shown to be important for CO2 conversion at high pressures. For fill pressures beyond 10 bar, CO2 may locally transit into supercritical states. The supercritical plasma kinetics may bypass atomic oxygen pathways and directly convert CO2 into O2. This work provides a detailed analysis into plasma-based high-pressure CO2 conversion, which is of great relevance to future large-scale sustainable carbon capture, utilization, and storage (CCUS).
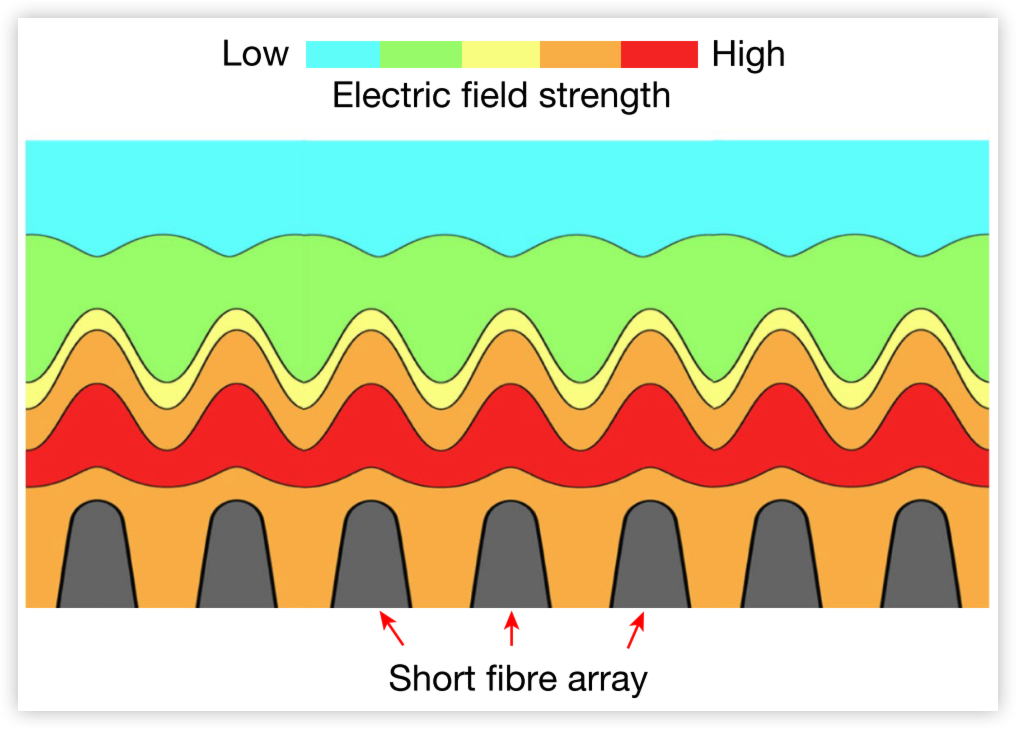
Plasmas can generate ultra-high-temperature reactive environments that can be used for the synthesis and processing of a wide range of materials. However, the limited volume, instability and non-uniformity of plasmas have made it challenging to scalably manufacture bulk, high-temperature materials. Here we present a plasma set-up consisting of a pair of carbon-fibre-tip-enhanced electrodes that enable the generation of a uniform, ultra-high temperature and stable plasma (up to 8,000 K) at atmospheric pressure using a combination of vertically oriented long and short carbon fibres. The long carbon fibres initiate the plasma by micro-spark discharge at a low breakdown voltage, whereas the short carbon fibres coalesce the discharge into a volumetric and stable ultra-high-temperature plasma. As a proof of concept, we used this process to synthesize various extreme materials in seconds, including ultra-high-temperature ceramics (for example, hafnium carbonitride) and refractory metal alloys. Moreover, the carbon-fibre electrodes are highly flexible and can be shaped for various syntheses. This simple and practical plasma technology may help overcome the challenges in high-temperature synthesis and enable large-scale electrified plasma manufacturing powered by renewable electricity.
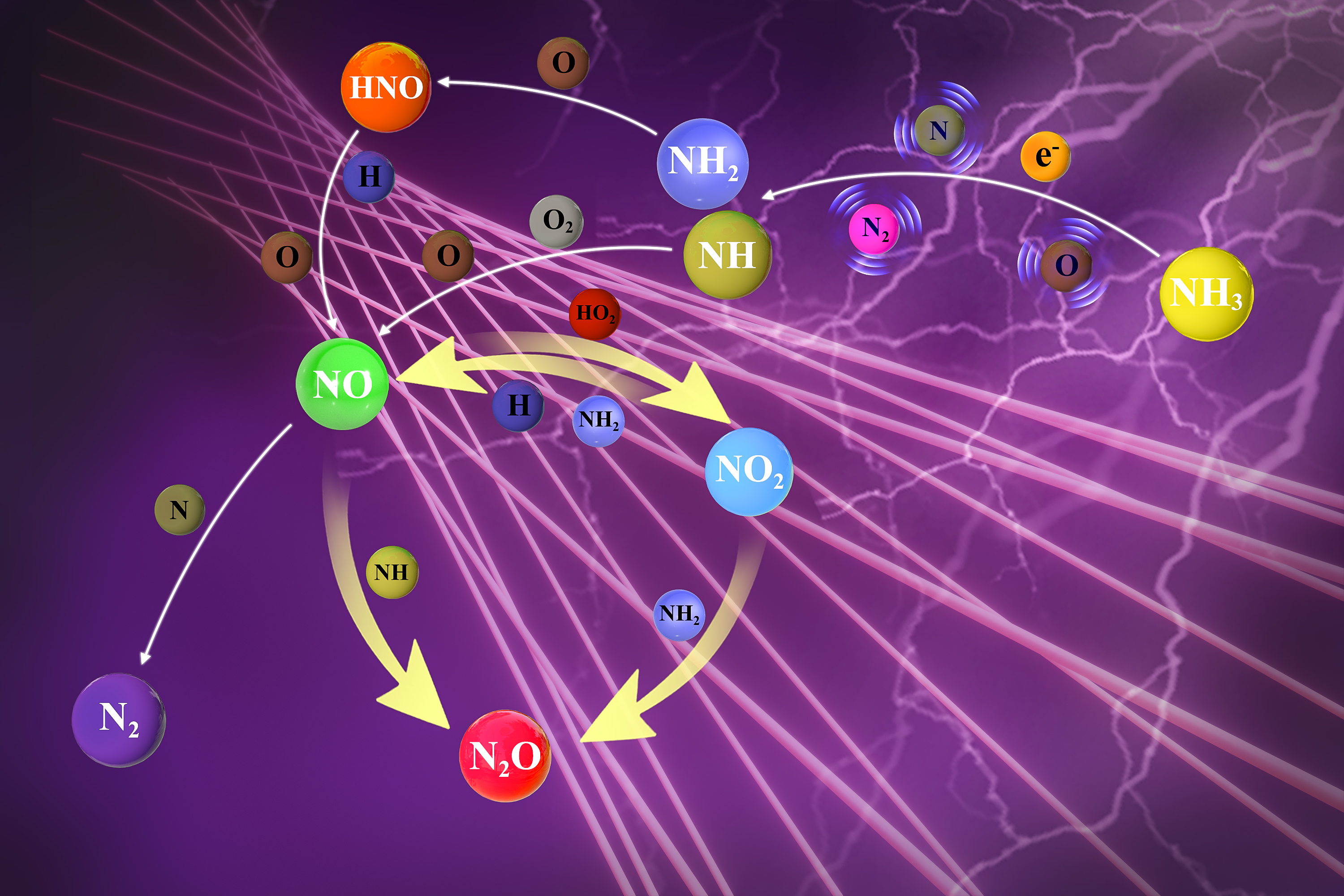
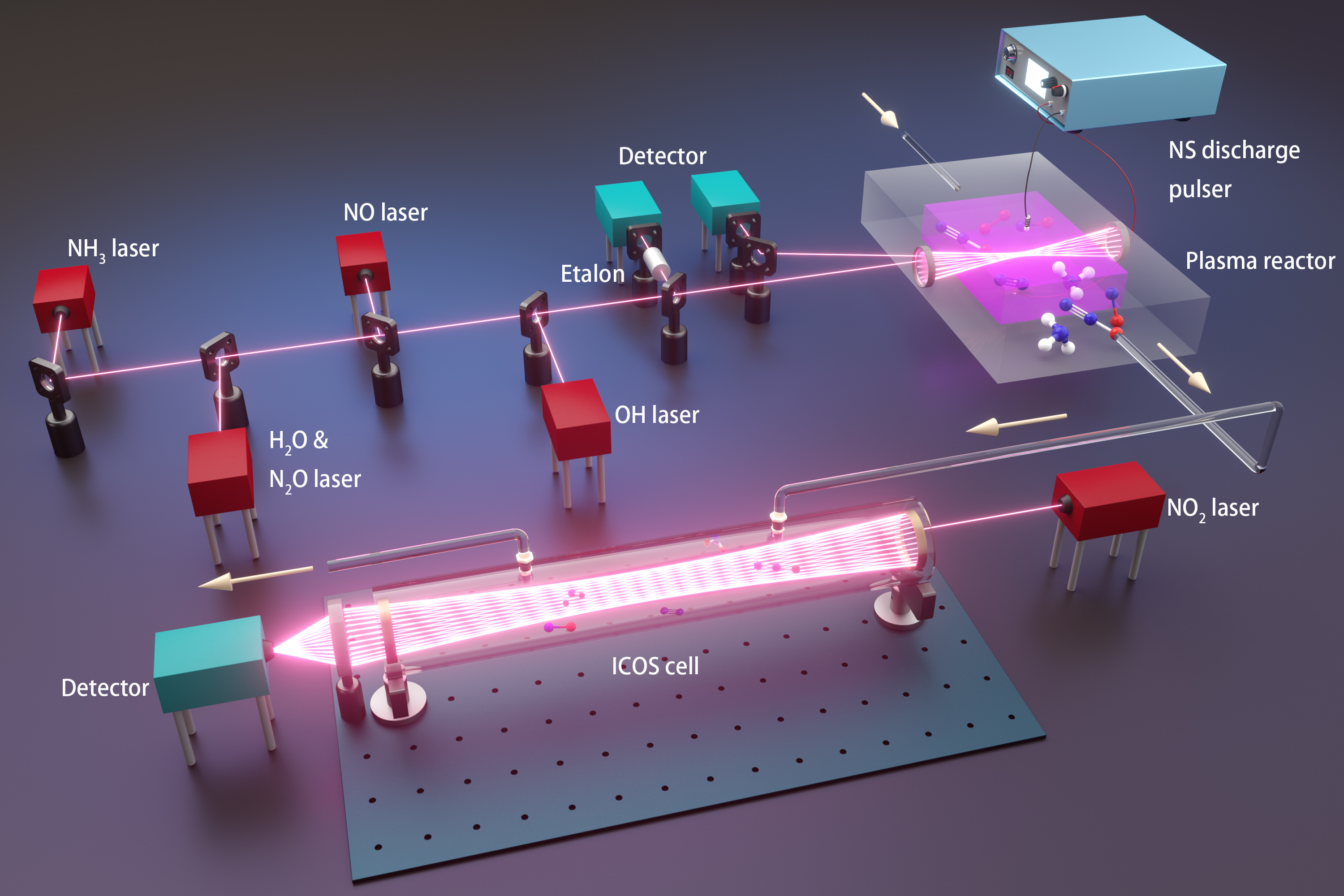
Ammonia represents one of most promising zero-carbon energy solutions to address the increasingly urgent climate change problem. However, the existing utilization of ammonia faces significant challenges from low chemical reactivity and high NOx/N2O emissions, which cause severe inefficiency issues such as cold start and cycle variability of power generation, and detrimental effects on air quality, respectively. A sustainable and energy-efficient approach to tackle the above challenges is low temperature plasma which enables non-equilibrium energy and chemical utilization of ammonia such as oxidation using renewable electricity. As such, this work, for the first time, explores plasma assisted ammonia oxidation at room temperature with a focus on unveiling non-equilibrium NOx/N2O reaction pathways by combining in situ laser diagnostics with plasma modeling. We found that the non-equilibrium plasma controls the NOx formation by supplying O/H/N atoms via electron-impact dissociation and collisional quenching of excited species. The N2O formation follows a two-step mechanism, where electron-impact reactions first provide amine radicals which further react with NOx to generate N2O. These pathways facilitate a high-efficiency and environment-friendly operation of plasma assisted ammonia oxidation with enhanced reactivity and reduced NOx/N2O emissions through manipulating mixture compositions and plasma discharge parameters.
The present study investigates the kinetics of low-temperature pyrolysis and oxidation of n-dodecane/O2/N2 mixtures in a repetitively-pulsed nanosecond discharge experimentally and numerically. Time-resolved TDLAS measurements, steady-state gas chromatography (GC) sampling, and mid-IR dual- modulation Faraday rotation spectrosocpy (DM-FRS) measurements are conducted to quantify temperature as well as species formation and evolution. A plasma-assisted n-dodecane pyrolysis and oxidation kinetic model incorporating the reactions involving electronically excited species and NOx chemistry is developed and validated. The results show that a nanosecond discharge can dramatically accelerate n-dodecane pyrolysis and oxidation at low temperatures. The numerical model has a good agreement with experimental data for the major intermediate species. From the pathway analysis, electronically excited N2* plays an important role in n-dodecane pyrolysis and oxidation. The results also show that with addition of n-dodecane, NO concentration is reduced considerably, which suggests that there is a strong NO kinetic effect on plasma-assisted low-temperature combustion via NO-RO2 and NO2-fuel radical reaction pathways. This work advances the understandings of the kinetics of plasma-assisted low-temperature fuel oxidation in N2/O2 mixtures.
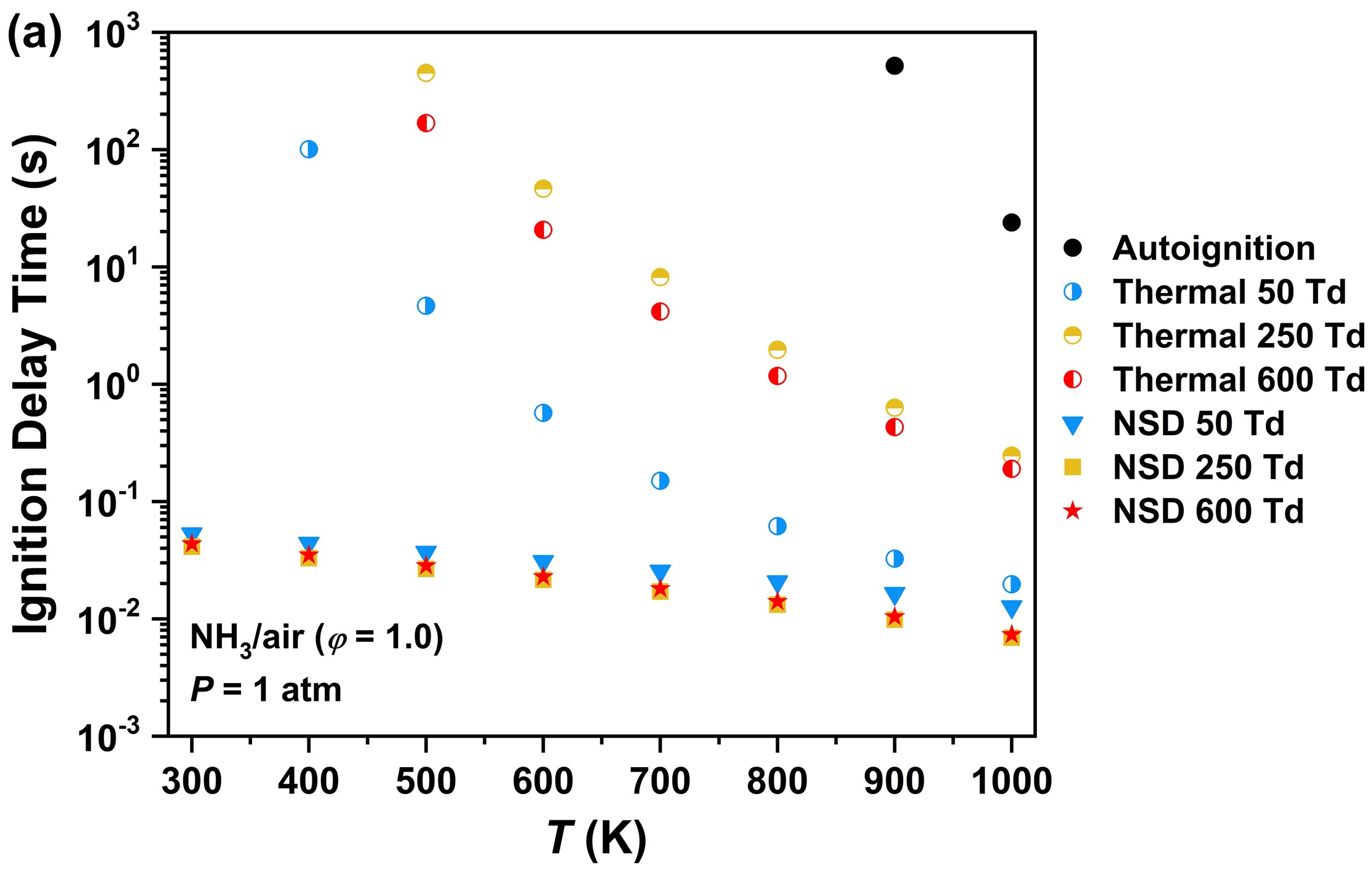
This work computationally studies plasma assisted low temperature NH3/air ignition and NOx formation in a repetitively-pulsed nanosecond discharge at atmospheric pressure by using an experimentally-validated plasma-combustion kinetic model. The results show that plasma discharge significantly enhances low temperature NH3 ignition. Compared with thermal ignition, the ignition delay time is shortened by 2-5 orders of magnitude due to the kinetic enhancement of excited species and radicals. The radicals (NH2, NH, H and O) produced through electron impact dissociation and quenching of electronically excited N2*, O(1D) and N(2D) promote OH production and further accelerate NH3 oxidation. The results show that there exists a non-monotonic dependence of ignition delay time on the reduced electric field strength. The optimum ignition enhancement is achieved at 250 Td at which the production of electronically excited species and radicals is most efficient. The vibrationally excited species produced at lower electric fields (<100 Td) are less effective in enhancing ignition because they only induce gas heating through the fast vibrational-translational relaxation by NH3 and H2O. At a higher electric field, although the efficient production of NH2, NH, H, O and OH by plasma creates new low temperature reaction pathways in enhancing low temperature NH3 ignition, the ignition is inhibited through NH + NO = N2O + H and chain-termination reaction NH2 + HO2 = NH3 + O2. The ignition delay times at different equivalence ratios show that the ignition enhancement by plasma is more effective at fuel-lean conditions due to the faster generation of N2(B), O(1D) and O from air, leading to accelerated NH3 oxidation via O(1D) + NH3 = NH2 + OH, NH3 + O = NH2 + OH and NH2 + O = NH + OH. The sensitivity analysis shows that the reactions involving O and O(1D) production are more effective on NH3 ignition enhancement than the fuel dissociation by electrons. Moreover, the ignition is also enhanced by NOx formation in plasma via reactions NH2 + NO = NNH + OH and NO + HO2 = NO2 + OH. This work advances the understanding of non-equilibrium excitation and NOx formation by plasma discharge on low temperature NH3 ignition.
This work numerically investigates the effects of non-equilibrium nanosecond plasma discharge pulse repetition frequency, pulse number, and flow velocity on the critical ignition volume, minimum ignition energy, and chemistry in a plasma-assisted H2/air flow at 300 K and 1 atm using a multi-scale adaptive reduced chemistry solver for plasma assisted combustion (MARCS-PAC). The interactions between discharges/ignition kernels spanning decoupled, partially-coupled and fully-coupled regimes in a pulse train are studied. For a single pulse discharge, increased flow velocity increases the minimum ignition energy required due to the increase of convective heat loss and flame stretch. The results show that the minimum ignition kernel propagation speed at the critical ignition kernel volume increases with the flow velocity. The minimum critical ignition volume decreases with the increase of plasma discharge energy. For sequential two-pulse discharges, ignition fails at both decoupled and partially-coupled regimes even when the total discharge energy is above the minimum ignition energy, but succeeds only in the fully-coupled regime at a shorter inter-pulse time. Overlap of the OH radical pool between the sequential two-pulse discharges and the increase of the chemistry effect due to the increase of reduced electric field in the fully-coupled regime contribute to the ignition enhancement. In addition, for two-pulse discharges in the fully-coupled discharge regime, the mixture can be ignited at a total energy below the minimum ignition energy of a single pulse with the same flow conditions. Moreover, for a given total discharge energy with multiple pulsed discharges, the enhancement of the ignition kernel volume has a non-monotonic dependence on discharge frequency and pulse number. The effective ignition enhancement can be achieved with an optimal pulse repetition frequency and pulse number. This work provides a new understanding of the mechanism for repetitive plasma ignition and insights for the optimization of plasma ignition in a reactive flow.
This work presents the results of two-dimensional modeling of the effects of non-equilibrium excitation and electrode geometry on H2/air ignition in a nanosecond plasma discharge. A multiscale adaptive reduced chemistry solver for plasma assisted combustion (MARCS-PAC) based on PASSKEy discharge modeling package and compressible multi-component reactive flow solver ASURF+ is developed and validated. This model is applied to simulate the impact of non-equilibrium plasma excitation and electrode geometry and heat loss on the dynamics of the discharge from streamer to spark and ignition kernel development in a H2/air mixture with a pair of cylindrical electrodes. The results show that the plasma-generated species (N2(A), N2(B), N2(a), N2(C), O(1D), O and H) in the spark and afterglow significantly accelerate the ignition kernel development. The increase of discharge voltage at the same total discharge energy promotes the non-equilibrium active species production. It is found that the production of electronically excited species at higher reduced electric field strength is more efficient in enhancing ignition in comparison to the vibrational excitation and heating. Moreover, the 2D simulation clearly reveals that the electric field and active species distribution are highly non-uniform. The streamers are initiated at the sharp outer edges of the negative and positive electrodes by a strong electric field while the electric field is much weaker at the centerline of the electrodes. Furthermore, the simulations reveal that the ignition enhancement is sensitive to the variation of electrode shape, diameter, and gap size due to the changes of electric field distribution and location of streamer formation. A cylindrical electrode produces a larger discharge volume and ignition kernel than the parabolic and spherical electrodes, when the discharge is localized near the axis of the gap. It is found that there is a non-monotonic dependence of ignition kernel size on the electrode diameter and inter-electrode distance. The increase of electrode diameter and gap size above the optimal conditions leads to the reduction of ignition kernel volume, due to the decrease of active species concentration and gas temperature. At a larger electrode surface area and electrode diameter as well as smaller electrode gap size, the heat loss to electrode plays a greater role in reducing the ignition kernel size and slowing ignition kernel development. This work provides insights and guidance to understand the kinetic enhancement of non-equilibrium plasma and the effects of electrode geometries on ignition for the optimization ignitors in advanced engines.
Layered nickel-rich materials are promising next-generation cathode materials for lithium ion batteries due to their high capacity and low cost. However, the poor thermal stability and longtime cycling performance hinders the commercial applications of high nickel materials. Doping with heteroatoms has been an effective approach for improving electrochemical performance of cathode materials. Controlling doping concentration and geometrical distribution is desired for optimal electrochemical performance, but it is challenging in traditional co-precipitation methods. In this work, controlled dysprosium (Dy) doping to NCM811 was studied in an aerosol synthesis method by controlling the precursor concentrations and heating parameters. The obtained materials were characterized by SEM, XRD, and XPS, and their electrochemical properties and thermal stability were evaluated. By controlling the doping concentration (1.5%), Dy-doped NCM811 was improved simultaneously in long-term cycling and high-rate performance. The thermal-chemical stability of the Dy-doped cathode materials was examined in a microflow reactor with a mass spectrometer. The results showed that Dy-doping shifted the O2 onset temperature to a higher temperature and reduced O2 release by 80%, thus dramatically increasing the thermal-chemical stability and improving the fire safety of cathode materials. Since high temperature aerosol synthesis is a low-cost and scalable method, the findings in this work have broad implications for commercial synthesis of novel materials with controlled doping modification to achieve high electrochemical performance and safety in lithium ion batteries.
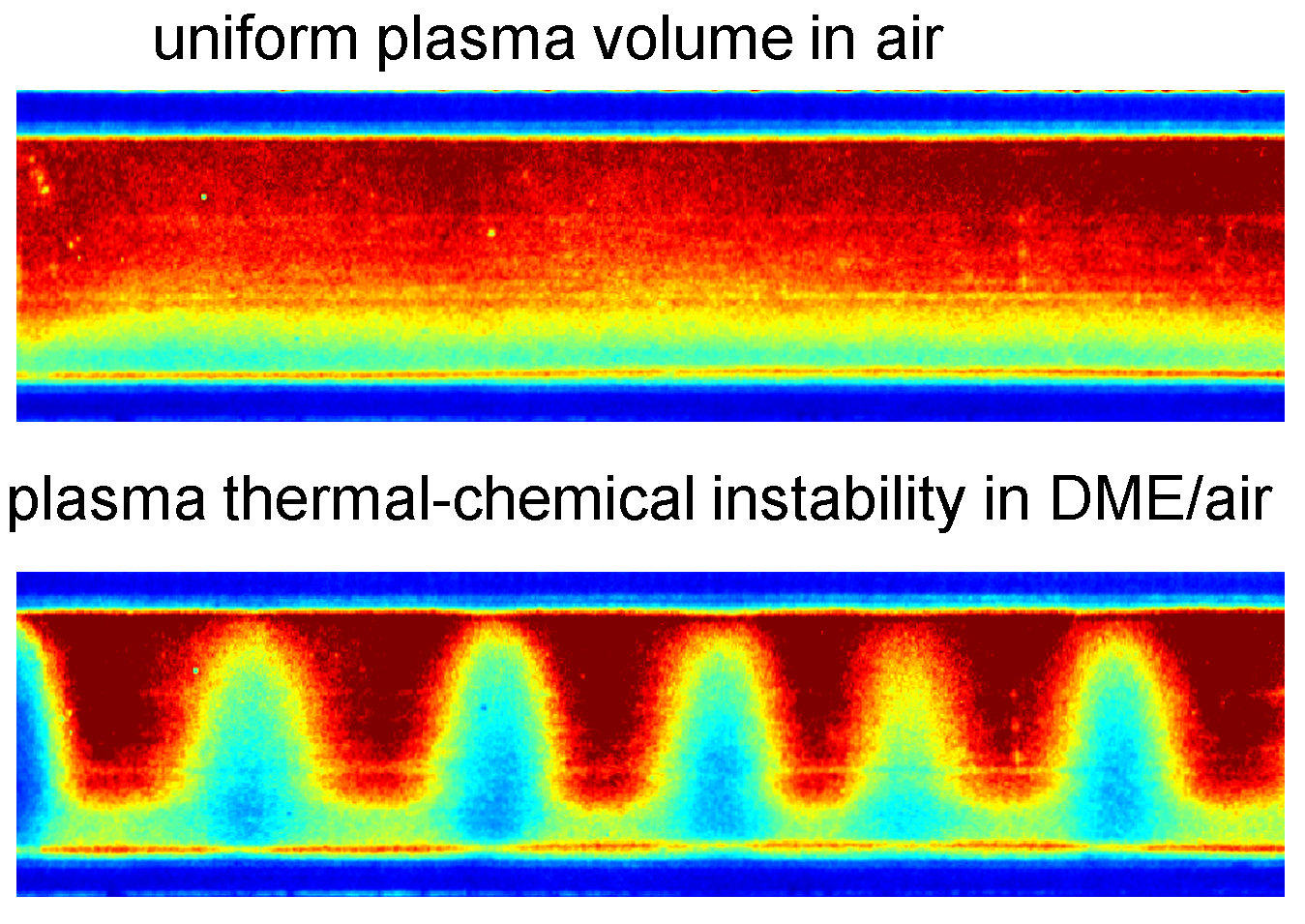
Plasma stability in reactive mixtures is critical for various applications from plasma-assisted combustion to gas conversion. To generate stable and uniform plasmas and control the transition towards filamentation, the underlying physics and chemistry need a further look. This work investigates the plasma thermal-chemical instability triggered by dimethyl-ether (DME) low-temperature oxidation in a repetitive nanosecond pulsed dielectric barrier discharge. First, a plasma-combustion kinetic mechanism of DME/air is developed and validated using temperature and ignition delay time measurements in quasi-uniform plasmas. Then the multi-stage dynamics of thermal-chemical instability is experimentally explored: the DME/air discharge was initially uniform, then contracted to filaments, and finally became uniform again before ignition. By performing chemistry modeling and analyzing the local thermal balance, it is found that such nonlinear development of the thermal-chemical instability is controlled by the competition between plasma-enhanced low-temperature heat release and the increasing thermal diffusion at higher temperature. Further thermal-chemical mode analysis identifies the chemical origin of this instability as DME low-temperature chemistry. This work connects experiment measurements with theoretical analysis of plasma thermal-chemical instability and sheds light on future chemical control of the plasma uniformity.
The stability of the weakly ionized plasma and the transition from a stable homogeneous discharge to unstable filaments play an important role in gas laser physics, plasma-assisted combustion, chemical reforming, and material synthesis. Theoretical stability analysis and thermal-chemical mode analysis (TCMA) were performed to understand the mechanism of plasma thermal-chemical instability by using a zero-dimensional plasma system with both simplified and detailed chemical kinetics of H2/O2/N2 mixtures. The plasma dynamic and kinetic models accounted for multiple physical mechanisms in the chemically-reactive weakly ionized plasma, including ionization, attachment/detachment, recombination, vibrational and electronic energy relaxation, convective and diffusive species/heat removal, Joule heating, and detailed chemical kinetics. An analytical criterion and the explosive mode species pointers (SP) were formulated while the representative active species (RAS) were identified for different thermal-chemical modes. The results showed that in addition to the classical thermal-ionization mechanism, various chemical modes from chemical heat imbalance and elementary kinetics significantly modified the time dynamics and the stability of the weakly ionized plasma. The present analysis provides insights and guidance to control plasma instability using chemical kinetics.
The control of plasma instability of weakly ionized plasma in a reactive flow is of great importance in plasma-assisted combustion, catalysis, fuel reforming and material synthesis. In this work, we propose a new concept of plasma chemical instability and analyze the mechanisms and impact of plasma chemical instability on the transition from a uniform discharge to a contracted state of a self-sustained glow discharge in a reactive H2–O2–N2 mixture. A one-dimensional numerical model for plasma chemical instability was developed which accounted for convective heat loss, Joule heating of plasma, and major non-equilibrium plasma-assisted combustion kinetic pathways including electron-impact ionization, vibrational energy transfer, electron attachment, combustion reactions, and heat release. The results showed that plasma chemical instability significantly modified the onset plasma current of plasma thermal instability. Specifically, the critical condition of the instability transition was strongly influenced by electron-impact reactant ionization, electron attachment to oxygen, endothermic/exothermic chemical reactions, and the formation of reaction products.
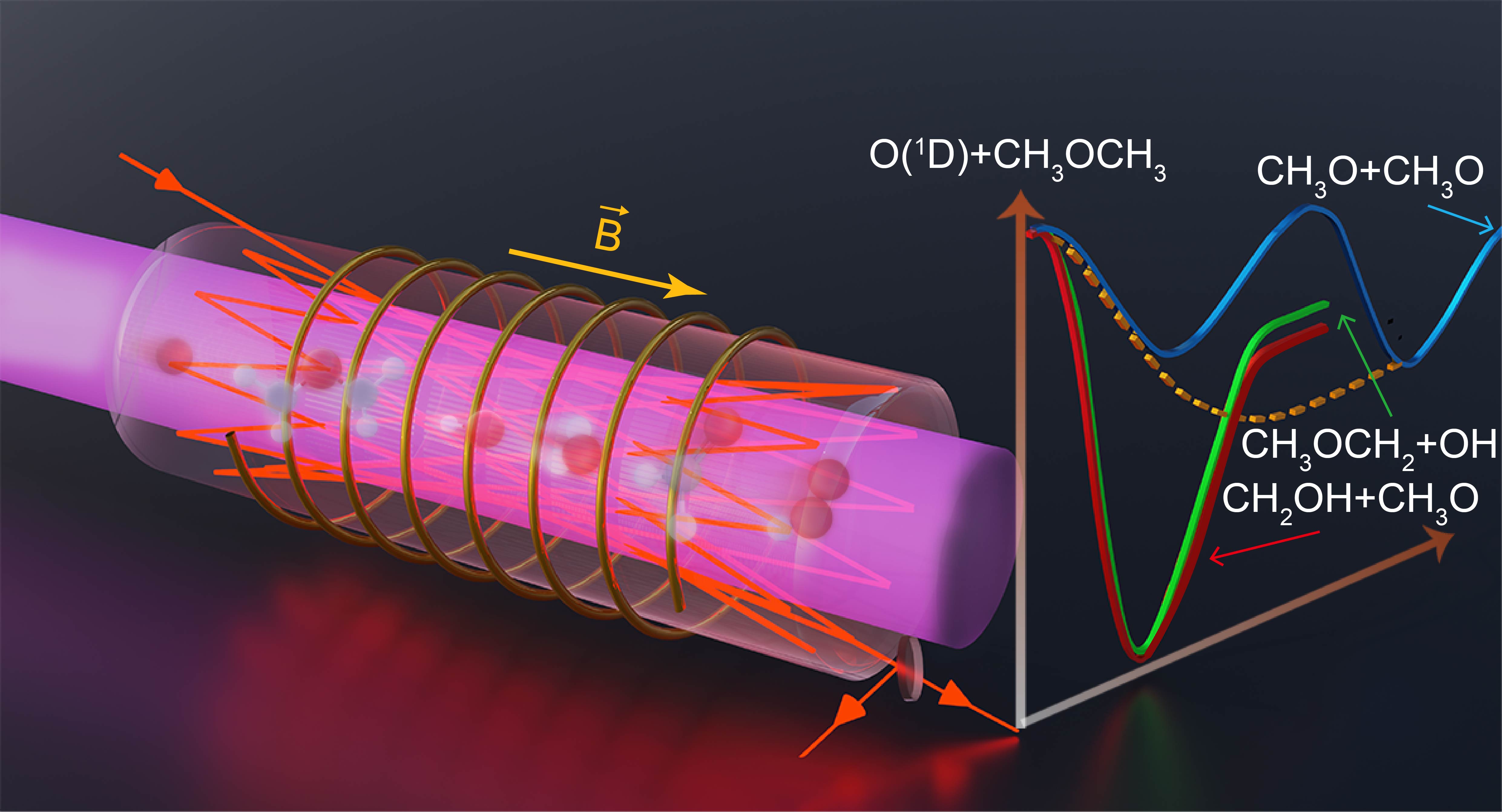
We combine in situ laser spectroscopy, quantum chemistry, and kinetic calculations to study the reaction of a singlet oxygen atom with dimethyl ether. Infrared laser absorption spectroscopy and Faraday rotation spectroscopy are used for the detection and quantification of the reaction products OH, H2O, HO2, and CH2O on submillisecond time scales. Fitting temporal profiles of products with simulations using an in-house reaction mechanism allows product branching to be quantified at 30, 60, and 150 Torr. The experimentally determined product branching agrees well with master equation calculations based on electronic structure data and transition state theory. The calculations demonstrate that the dimethyl peroxide (CH3OOCH3) generated via O-insertion into the C–O bond undergoes subsequent dissociation to CH3O + CH3O through energetically favored reactions without an intrinsic barrier. This O-insertion mechanism can be important for understanding the fate of biofuels leaking into the atmosphere and for plasma-based biofuel processing technologies.
The multi-channel reaction of excited singlet oxygen atom with ethanol, O(1D) + C2H5OH (1), was studied in a photolysis flow reactor coupled with mid-infrared Faraday rotation spectroscopy (FRS) and UV-IR direct absorption spectroscopy (DAS) at 297 K with reactor pressures of 60, 120 and 150 Torr (bath He). The excited singlet oxygen atom was generated through the photolysis of O3 at 266 nm. The photon flux and O(1D) concentrations were determined by in situ actinometry based on O3 depletion. Temporal profiles of OH and H2O were monitored via DAS signals at ca. 3568.62 cm-1 and 3568.29 cm-1, while temporal profiles of HO2 were measured via FRS signals at ca. 1396.90 cm-1. The branching ratios of the target reaction (1) were determined by fitting temporal profiles to simulations from an in-house reaction mechanism. Two major reaction channels were identified as CH3CHOH + OH and CH3O + CH2OH, and their branching ratios were determined as 0.46 ± 0.12 and 0.42 ± 0.11, respectively. A specific HO2 + RO2 reaction between HO2 and O2CH2CH2OH (β-RO2) at the low temperature range is estimated in this work as HO2 + O2CH2CH2OH -> products with a rate constant of 7 × 10-12 cm3 molecule-1 s-1.
The rate constant and branching ratios of ethyl reaction with hydroperoxyl radical, C2H5 + HO2 (1), a key radical-radical reaction for intermediate temperature combustion chemistry, were measured in situ for the first time in a photolysis Herriott cell by using mid-IR Faraday rotation spectroscopy (FRS) and UV-IR direct absorption spectroscopy (DAS). The microsecond time-resolved diagnostic technique in this work enabled the direct rate measurements of the target reaction at 40 and 80 mbar and reduced the experimental uncertainty considerably. C2H5 and HO2 radicals were generated by the photolysis of (COCl)2/C2H5I/CH3OH/O2/He mixture at 266 nm. By direct measurements of the transient profiles of C2H5, HO2 and OH concentrations, the overall rate constant for this reaction at 297 K was determined as k1(40 mbar) = (3.8 ± 0.8) × 10-11 cm3 molecule-1 s-1 and k1(80 mbar) = (4.1 ± 1.0) × 10-11 cm3 molecule-1 s-1 The direct observation of hydroxyl radical (OH) indicated that OH formation channel was the major channel with a branching ratio of 0.8 ± 0.1.
Non-equilibrium plasmas derive their low temperature reactivity from producing and driving energetic electrons and active species under large electric fields. Therefore, the impact of reactants on the plasma properties including electron number density, electric field, and electron temperature is critical for applications such as plasma CH4 reforming. Due to experimental complexity, electron properties and the electric field are rarely measured together in the same discharge. In this work, we combine time-resolved Thomson scattering and electric field induced second harmonic generation (EFISH) to probe electron temperature, electron density, and electric field strength in a 60 Torr CH4/Ar nanosecond-pulsed dielectric barrier discharge (ns-DBD) while varying the CH4 mole fraction from 0 to 8%. These measurements are compared to a 1-D numerical model to benchmark its predictions and identify areas of uncertainty. Nonlinear coupling between CH4 addition, electron temperature, electron density, and the electric field was directly observed. Contrary to previous measurements in He, the electron temperature increased with CH4 mole fraction. This rise in electron temperature is identified as electron heating by residual electric fields that increased with larger CH4 mole fraction. Moreover, the electron number density has been found to decrease rapidly with the increase of methane mole fraction. Comparison of these measurements with the model yielded better agreement at higher CH4 mole fractions and with the usage of ab initio calculated Ar electron-impact cross-sections from the B-spline R-matrix (BSR) database. Furthermore, the calculated plasma properties are shown to be sensitive to the residual surface charge implanted on the quartz dielectric surfaces. Without considering surface charge in the simulations, the calculated electric field profiles agreed well with the measurements, but the electron properties were underpredicted by more than a factor of three. Therefore, measurements of either the electric field or electron properties measurements alone are insuffcient to fully validate modelling predictions.
In many low temperature plasmas (LTPs), the OH radical and temperature represent key properties of plasma reactivity. However, OH and temperature measurements in weakly ionized LTPs are often challenging, due to the low concentration and short lifetime of OH and alsoand the abrupt temperature rise caused by fast gas heating. To address such issue, this letter combined cavity enhanced absorption spectroscopy (CEAS) with femtosecond (fs) pulses to enable sensitive single-shot broadband measurements of OH concentration and temperature with a time resolution of ∼ 180 nanoseconds (ns) in LTPs. Such combination leveraged several diagnostic benefits. With the appropriately designed cavity, an absorption gain of ∼ 66 was achieved, enhancing the actual OH detection limit by ∼ 55× to the 1011 cm3 level (sub-ppm in this work) compared to single-pass absorption. Single-shot measurements were enabled while maintaining a time resolution of ∼ 180 ns sufficiently short for detecting OH with a lifetime of ∼ 100 μs. With the broadband fs laser, ~34000 cavity modes were matched with ~95 modes matched on each CCD pixel bandwidth, such that the fs-CEAS signal became highly immune to the laser-cavity coupling noise and considerably more robust across the entire spectral range. Also, the broadband fs laser allowed simultaneous sensing of many absorption features, which were exploited to enable simultaneous multi-parameter measurements with enhanced accuracies.
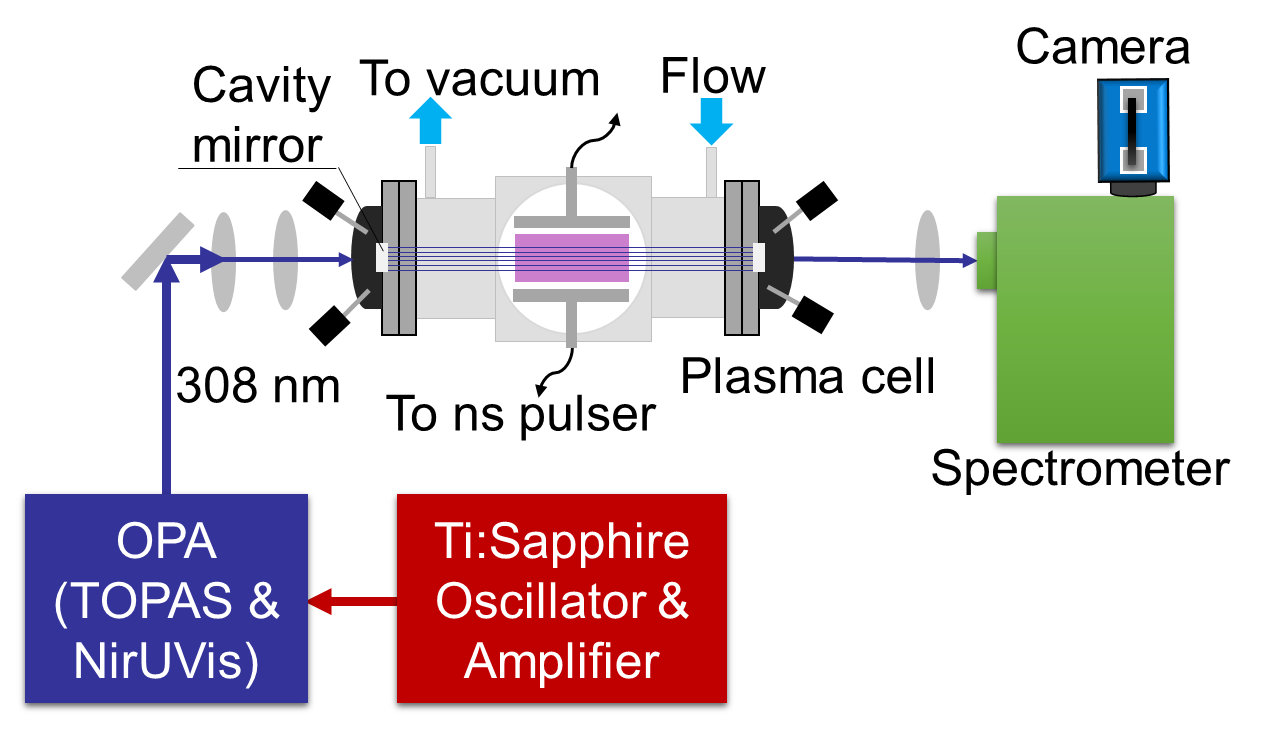
This Letter reports a femtosecond ultraviolet laser absorption spectroscopy (fs-UV-LAS) for simultaneous in situ measurements of temperature and species. This fs-UV-LAS technique was demonstrated based on X2Π-A2Σ+ transitions of OH radicals near 308 nm generated in low temperature plasmas and flames. The fs-UV-LAS technique has revealed three major diagnostic benefits. First, a series of absorption features within a spectral bandwidth of ∼ 3.2 nm near 308nm were simultaneously measured and then enabled simultaneous multi-parameter measurements with enhanced accuracy. The results show that the temperature and OH concentration could be measured with accuracy enhanced by 29-88% and 58–91%, respectively, compared to those obtained with past two-narrow-line absorption methods. Second, an ultrafast time resolution of ∼ 120 picoseconds was accomplished for the measurements. Third, due to the large OH X2Π-A2Σ+ transitions in the UV range, a simple single-pass absorption with a 3-cm path length was allowed for measurements in plasmas with low OH number density down to ∼ 2 × 1013 cm3. Also due to the large OH UV transitions, single-shot fs absorption measurements were accomplished in flames, which was expected to offer more insights into chemically reactive flow dynamics.
Faraday rotation spectroscopy (FRS) employs the Faraday effect to detect Zeeman splitting in the presence of a magnetic field. In this article, we present system design and implementation of radical sensing in a photolysis reactor using FRS. High sensitivity (100 ppb) and time resolved in situ HO2 detection is enabled with a digitally balanced acquisition scheme. Specific advantages of employing FRS for sensing in such dynamic environments are examined and rigorously compared to the more established conventional laser absorption spectroscopy (LAS). Experimental results show that FRS enables HO2 detection when LAS is deficient, and FRS compares favorably in terms of precision when LAS is applicable. The immunity of FRS to spectral interferences such as absorption of hydrocarbons and other diamagnetic species absorption and optical fringing are highlighted in comparison to LAS.
Understanding the multi-channel dynamics of O(1D) reactions with unsaturated hydrocarbon molecules in low temperature reaction kinetics is critically important in stratospheric chemistry, plasma chemistry, plasma assisted fuel reforming, materials synthesis, and plasma assisted combustion. A photolysis flow reactor coupled with highly selective mid-infrared Faraday Rotation Spectroscopy (FRS) and direct ultraviolet-infrared (UV-IR) absorption spectroscopy (DAS) techniques was developed for the first time to study the multi-channel dynamics of excited singlet oxygen atom O(1D) reactions with C2H2 and the kinetics of subsequent reactions. Time-resolved species concentrations of OH, HO2 and H2O were obtained and used to develop a validated kinetic model of O(1D) reactions with C2H2. The branching ratios of O(1D) reaction with C2H2 and subsequent HO2 kinetics were also quantified. It is found that, contrary to O(1D) reactions with saturated alkanes, OH formation via direct H abstraction by O(1D) is negligible. The results revealed that two chain-branching and propagation reactions via direct O(1D) insertion are the major pathways for radical production. The present study clearly demonstrated the advantage of radical detection and kinetic studies using FRS in the effective suppression of absorption interference from non-paramagnetic hydrocarbons.
We demonstrate the use of laser-induced incandescence (LII) of submicron tungsten carbide (WC) particles as a method for particle image velocimetry (PIV). The technique allows a single laser to be used for separate measurements of velocity of two phases in a droplet-laden flow. Submicron WC particles are intentionally seeded into a two-phase flow, and heated by a light sheet generated by a double-pulsed PIV laser operating at sufficiently high pulse energy. The small size and large absorption cross section allows particles to be heated up to several thousand degrees Kelvin to emit strong incandescence signals, whilst the laser-induced temperature increase in liquid droplets/large particles is negligible. The incandescence signal from WC and Mie scattering from droplets/large particles are separately captured by deploying different filters to a PIV camera. The consecutive images of the laser-induced incandescence (LII) are used to determine the velocity field of the gas-phase flow, and those of Mie scatter are used to extract the velocity of droplets/large particles. The proposed technique is demonstrated in an air jet first and compared with the result given by a normal PIV test, which shows that submicron WC particles can accurately follow the gas flow, and that the LII images can be used to perform cross-correlations. We then apply this technique on an ethanol droplet/air jet (non-reacting), demonstrating the resulting slip velocity between two phases. The proposed technique combining PIV and LII with a single laser requires little additional equipment, and is applicable to a much higher droplet/particle density than previously feasible. Finally, the possibility of applying this technique to a flame is demonstrated and discussed.
We report measurements of HO2 radicals using wavelength modulated Faraday Rotation Spectroscopy in a multi-pass photolysis reactor. The current setup enables line-locked measurements to improve sensitivity and time resolution compared to the line-scanning system.
"Research as Art, Art as Research". More in Hongtao's Gallery.
Working Paper in AI4Science
Gaussian-Process Modeling of Turbulent Viscosity in Turbulent Boundary Layers
Recently great attention in the turbulence modeling community has been drawn to data-driven methods. Along with sophisticated statistical machine learning algorithms and the availability of big data, a new era of “black-box” turbulence models is coming, targeting at better tuning and more detailed uncertainty quantification. In this work, a Gaussian process (GP) regression is proposed for modeling the turbulent viscosity (T-V), which has been discussed for decades. However, current T-V modeling is mostly parametric with poor validity of model parameters reported in various turbulent flows. The proposed non-parametric GP model is trained and tested by high-fidelity data collected from direct numerical simulations (DNS). Both geometry-dependent and physics-informed features are discussed and R2 is over 0.85 for both cases. Additionally, the GP model trained by incompressible boundary layer flows is shown to have the capability to predict T-Vs in compressible flows and general wall flows with matching Reynolds number and proper scaling conversion. More validation cases involving DNS data and RANS simulations are required to better characterize this approach. Ultimately, this non-parametric data-driven method will allow for a more efficient and better uncertainty quantified model of turbulent viscosity and Reynolds stress.
Learning-based Reconstruction of Chemical Reaction Networks
Dynamics of chemical reaction networks (CRNs) are of central importance for understanding various phenomena including explosion, catalysis, and biological systems. Traditionally, the determination of rate constants of individual reactions in CRNs requires much prior knowledge, complicated numerical integration, and contains large uncertainty. In this work, we propose to apply machine learn- ing techniques including linear regression, recurrent neural network (RNN), and Gaussian process (GP) model to determine the reaction rate constants and recon- struct chemical reaction networks from discrete experimental data. We showed that RNN best reserved the system dynamics from small to large-scale systems, while linear regression with poly-terms performed best in simple kinetics but failed to capture nonlinear dynamics in real systems. With the promising per- formance of RNN in fully-observed systems, our work can be further applied and tested in partially-observed nonlinear systems, potentially facilitating research in complex chemical kinetics.